Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to PADCEV 1.25 mg/kg in combination with intravenous pembrolizumab for the treatment of MIBC in 167 patients in EV-303 (NCT03924895) and for the treatment of locally advanced or mUC in 564 patients in EV-302 (NCT04223856) and EV-103 (NCT03288545); PADCEV as a single agent at 1.25 mg/kg in 720 patients in EV-301 (NCT03474107), EV-201 (NCT03219333), EV-203 (NCT04995419), EV-101 (NCT02091999), and EV-102 (NCT03070990). Ocular disorders reflect 384 patients in EV‑201, EV-101, and EV-102.
Among 167 patients receiving PADCEV in combination with intravenous pembrolizumab for the treatment of MIBC, the most common (≥20%) adverse reactions, including laboratory abnormalities, were increased glucose, decreased hemoglobin, increased aspartate aminotransferase, rash, increased alanine aminotransferase, fatigue, pruritus, increased creatinine, decreased sodium, decreased lymphocytes, peripheral neuropathy, increased potassium, alopecia, dysgeusia, diarrhea, decreased appetite, constipation, nausea, decreased phosphate, urinary tract infection, dry eye, and decreased weight.
Among 564 patients receiving PADCEV in combination with intravenous pembrolizumab for the treatment of locally advanced or mUC, 59% were exposed to PADCEV for ≥6 months, and 24% were exposed for ≥12 months. In this pooled population, the most common (≥20%) adverse reactions, including laboratory abnormalities, were increased aspartate aminotransferase, increased creatinine, rash, increased glucose, peripheral neuropathy, increased lipase, decreased lymphocytes, increased alanine aminotransferase, decreased hemoglobin, fatigue, decreased sodium, decreased phosphate, decreased albumin, pruritus, diarrhea, alopecia, decreased weight, decreased appetite, increased urate, decreased neutrophils, decreased potassium, dry eye, nausea, constipation, increased potassium, dysgeusia, urinary tract infection, and decreased platelets.
Among 720 patients receiving PADCEV as a single agent, 37% were exposed for ≥6 months, and 14% were exposed for ≥12 months. In this pooled population, the most common (≥20%) adverse reactions, including laboratory abnormalities, were increased glucose, increased aspartate aminotransferase, decreased lymphocytes, increased creatinine, rash, fatigue, peripheral neuropathy, decreased albumin, decreased hemoglobin, alopecia, decreased appetite, decreased neutrophils, decreased sodium, increased alanine aminotransferase, decreased phosphate, diarrhea, nausea, pruritus, increased urate, dry eye, dysgeusia, constipation, increased lipase, decreased weight, decreased platelets, abdominal pain, and dry skin.
The data described in the following section reflects exposure to PADCEV in combination with intravenous pembrolizumab from EV‑302, the dose escalation cohort, Cohort A and Cohort K of EV-103, and EV-303. Patients received PADCEV 1.25 mg/kg in combination with intravenous pembrolizumab until disease progression or unacceptable toxicity.
The data described in the following section also reflects exposure to PADCEV as a single agent from an open-label, randomized, trial (EV‑301) and Cohort 1 and Cohort 2 of an open-label, single arm, two cohort trial (EV-201). Patients received PADCEV 1.25 mg/kg until disease progression or unacceptable toxicity.
Neoadjuvant and Adjuvant Treatment of Cisplatin-Ineligible Patients with MIBC
EV-303
The safety of PADCEV in combination with intravenous pembrolizumab as neoadjuvant treatment and continued after radical cystectomy (RC) as adjuvant treatment was evaluated in an open-label, randomized, multicenter trial (EV-303) in patients with previously untreated MIBC who were ineligible for or declined cisplatin-based chemotherapy. Patients received PADCEV 1.25 mg/kg in combination with intravenous pembrolizumab (n=167) before and after RC with pelvic lymph node dissection (PLND) or RC with PLND alone (n=159)
For the 167 patients who received PADCEV in the neoadjuvant phase, the median duration of exposure to PADCEV was 1.6 months (range: 0.03 to 2.8 months) and the median number of cycles of PADCEV was 3 (range: 1, 3) in the neoadjuvant phase. For the 92 patients who received PADCEV in the adjuvant phase, the median duration of exposure to PADCEV was 3.7 months (range: 0.03 to 7.6 months) and the median number of cycles of PADCEV was 6 (range: 1, 6) in the adjuvant phase. Across the combined neoadjuvant and adjuvant phases (n=167), the median number of cycles of PADCEV was 5 (range: 1, 9) out of a planned 9 cycles.
Table 5 summarizes the most common (≥20%) adverse reactions in EV-303. Clinically relevant adverse reactions (<20%) include dry skin (15%), hypothyroidism (14%), vomiting (9%), pneumonitis/ILD (4.2%), skin hyperpigmentation (3%), infusion site extravasation (1.2%), and myasthenia gravis and myositis (0.6% each).
Neoadjuvant Phase of EV-303
A total of 167 patients received at least one dose of PADCEV in combination with intravenous pembrolizumab as neoadjuvant treatment before receiving RC.
In the neoadjuvant phase, serious adverse reactions occurred in 27% of patients receiving PADCEV in combination with intravenous pembrolizumab. The most frequent (≥2%) serious adverse reactions were urinary tract infection (3.6%) and hematuria (2.4%). Fatal adverse reactions occurred in 1.2% of patients including myasthenia gravis and toxic epidermal necrolysis (0.6% each). Additional fatal adverse reactions were reported in 2.7% of patients in the post-surgery phase before adjuvant treatment started, including sepsis and intestinal obstruction (1.4% each).
Adverse reactions leading to discontinuation of PADCEV in the neoadjuvant phase occurred in 22% of patients. The most common adverse reactions (≥1%) leading to discontinuation of PADCEV were rash (4.8%), peripheral neuropathy (2.4%), and diarrhea, dysgeusia, fatigue, pruritus, and toxic epidermal necrolysis (1.2% each).
Adverse reactions leading to dose interruption of PADCEV in the neoadjuvant phase occurred in 29% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were rash (8%), neutropenia (3.6%), hyperglycemia (3%), and fatigue and peripheral neuropathy (2.4% each).
Adverse reactions leading to dose reduction of PADCEV in the neoadjuvant phase occurred in 13% of patients. The most common adverse reactions (≥1%) leading to dose reduction of PADCEV were rash (4.8%), pruritus (1.8%), and peripheral neuropathy, increase alanine aminotransferase, increased aspartate aminotransferase, decreased appetite, fatigue, neutropenia, and decreased weight (1.2% each).
Of the 167 patients in the PADCEV in combination with intravenous pembrolizumab arm who received neoadjuvant treatment, 7 (4.2%) patients did not receive surgery due to adverse reactions. The adverse reactions that led to cancellation of surgery were acute myocardial infarction, bile duct cancer, colon cancer, respiratory distress, urinary tract infection and the deaths due to myasthenia gravis and toxic epidermal necrolysis (0.6% each).
Of the 146 patients who received neoadjuvant treatment with PADCEV in combination with intravenous pembrolizumab and underwent RC, 6 (4.1%) patients experienced delay of surgery (defined as time from last neoadjuvant treatment to surgery exceeding 8 weeks) due to adverse reactions.
Adjuvant Phase of EV-303
Patients who did not proceed to surgery were ineligible for adjuvant treatment. Of the 149 patients who underwent surgery, 100 patients received adjuvant treatment with PADCEV in combination with intravenous pembrolizumab. Of the 49 patients who did not receive adjuvant treatment, discontinuation of treatment with PADCEV in combination with intravenous pembrolizumab prior to the adjuvant phase was due to an adverse event in 21 patients.
In the adjuvant phase, serious adverse reactions occurred in 43% of patients receiving PADCEV in combination with intravenous pembrolizumab. The most frequent (≥2%) serious adverse reactions were urinary tract infection (8%), acute kidney injury and pyelonephritis (5% each), urosepsis (4%), and hypokalemia, intestinal obstruction, and sepsis (2% each). Fatal adverse reactions occurred in 7% of patients, including urosepsis, hemorrhage intracranial, death, myocardial infarction, multiple organ dysfunction syndrome, and pneumonia pseudomonal (1% each).
Adverse reactions leading to discontinuation of PADCEV in the adjuvant phase occurred in 26% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were peripheral neuropathy (5%) and rash (4%).
Adverse reactions leading to dose interruption of PADCEV in the adjuvant phase occurred in 36% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were rash (6%), diarrhea and urinary tract infection (5% each), fatigue (4%), pruritus (3%), and peripheral neuropathy and pyelonephritis (2% each).
Adverse reactions leading to dose reduction of PADCEV in the adjuvant phase occurred in 7% of patients. The most common adverse reaction (≥2%) leading to dose reduction of PADCEV was weight decreased (2%).
Previously Untreated Locally Advanced or mUC
EV-302
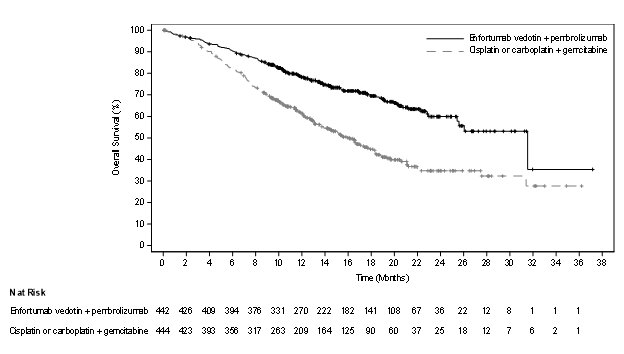
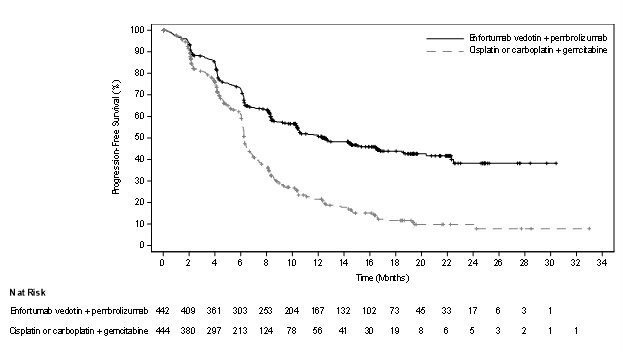
The safety of PADCEV in combination with intravenous pembrolizumab was evaluated in an open-label, randomized, multicenter trial (EV-302) in patients with locally advanced or mUC. Patients received either PADCEV 1.25 mg/kg and pembrolizumab (n=440) or gemcitabine and platinum chemotherapy (either cisplatin or carboplatin) (n=433). Among patients who received PADCEV and pembrolizumab, the median duration of exposure for PADCEV was 7 months (range: 0.3 to 31.9 months).
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with intravenous pembrolizumab. The most common serious adverse reactions (≥2%) were rash (6%), acute kidney injury (5%), pneumonitis/ILD (4.5%), urinary tract infection (3.6%), diarrhea (3.2%), pneumonia (2.3%), pyrexia (2%), and hyperglycemia (2%).
Fatal adverse reactions occurred in 3.9% of patients treated with PADCEV in combination with intravenous pembrolizumab including acute respiratory failure (0.7%), pneumonia (0.5%), and pneumonitis/ILD (0.2%).
Adverse reactions leading to discontinuation of PADCEV occurred in 35% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were peripheral neuropathy (15%), rash (4.1%), and pneumonitis/ILD (2.3%).
Adverse reactions leading to dose interruption of PADCEV occurred in 73% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were peripheral neuropathy (22%), rash (16%), COVID-19 (10%), diarrhea (5%), pneumonitis/ILD (4.8%), fatigue (3.9%), hyperglycemia (3.6%), increased alanine aminotransferase (3%), and pruritus (2.5%).
Adverse reactions leading to dose reduction of PADCEV occurred in 42% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were rash (16%), peripheral neuropathy (13%), and fatigue (2.7%).
Table 7 summarizes the most common (≥15%) adverse reactions in EV-302. Clinically relevant adverse reactions (<15%) include vomiting (12%), pneumonitis/ILD and hypothyroidism (10% each), blurred vision and skin hyperpigmentation (6% each), infusion site extravasation (1.8%), and myositis (0.5%).
Previously Untreated Cisplatin-Ineligible Patients with Locally Advanced or mUC
EV-103
The safety of PADCEV was evaluated in combination with intravenous pembrolizumab in a multi cohort trial (EV-103) in 121 patients with locally advanced or mUC who were not eligible for cisplatin-containing chemotherapy and received at least one dose of PADCEV 1.25 mg/kg and pembrolizumab
Serious adverse reactions occurred in 50% of patients treated with PADCEV in combination with intravenous pembrolizumab. The most common serious adverse reactions (≥2%) were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), sepsis (3.3%), pneumonia (3.3%), hematuria (3.3%), pneumonitis/ILD (3.3%), urinary retention (2.5%), diarrhea (2.5%), myasthenia gravis (2.5%), myositis (2.5%), anemia (2.5%), and hypotension (2.5%).
Fatal adverse reactions occurred in 5% of patients treated with PADCEV in combination with intravenous pembrolizumab including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis/ILD (0.8%).
Adverse reactions leading to discontinuation of PADCEV occurred in 36% of patients. The most common adverse reactions (≥2%) leading to discontinuation of PADCEV were peripheral neuropathy (20%) and rash (6%).
Adverse reactions leading to dose interruption of PADCEV occurred in 69% of patients. The most common adverse reactions (≥2%) leading to dose interruption of PADCEV were peripheral neuropathy (18%), rash (12%), increased lipase (6%), pneumonitis/ILD (6%), diarrhea (4.1%), acute kidney injury (3.3%), increased alanine aminotransferase (3.3%), fatigue (3.3%), neutropenia (3.3%), urinary tract infection (3.3%), increased amylase (2.5%), anemia (2.5%), COVID-19 (2.5%), hyperglycemia (2.5%), and hypotension (2.5%).
Adverse reactions leading to dose reduction of PADCEV occurred in 45% of patients. The most common adverse reactions (≥2%) leading to dose reduction of PADCEV were peripheral neuropathy (17%), rash (12%), fatigue (5%), neutropenia (5%), and diarrhea (4.1%).
Table 9 summarizes the most common (≥20%) adverse reactions in EV-103. Clinically relevant adverse reactions (<20%) include vomiting (20%), pyrexia (18%), hypothyroidism (11%), pneumonitis/ILD (10%), skin hyperpigmentation (8%), myasthenia gravis (2.5%), myositis (3.3%), and infusion site extravasation (0.8%).
Previously Treated Locally Advanced or mUC
EV-301
The safety of PADCEV was evaluated as a single agent in EV-301 in patients with locally advanced or mUC (n=296) who received at least one dose of PADCEV 1.25 mg/kg and who were previously treated with a PD-1 or PD-L1 inhibitor and a platinum-based chemotherapy
Serious adverse reactions occurred in 47% of patients treated with PADCEV. The most common serious adverse reactions (≥2%) were urinary tract infection, acute kidney injury (7% each), and pneumonia (5%). Fatal adverse reactions occurred in 3% of patients, including multiorgan dysfunction (1%), hepatic dysfunction, septic shock, hyperglycemia, pneumonitis/ILD, and pelvic abscess (0.3% each).
Adverse reactions leading to discontinuation occurred in 17% of patients; the most common adverse reactions (≥2%) leading to discontinuation were peripheral neuropathy (5%) and rash (4%).
Adverse reactions leading to dose interruption occurred in 61% of patients; the most common adverse reactions (≥4%) leading to dose interruption were peripheral neuropathy (23%), rash (11%), and fatigue (9%).
Adverse reactions leading to dose reduction occurred in 34% of patients; the most common adverse reactions (≥2%) leading to dose reduction were peripheral neuropathy (10%), rash (8%), decreased appetite (3%), and fatigue (3%).
Table 11 summarizes the most common (≥15%) adverse reactions in EV-301. Clinically relevant adverse reactions (<15%) include vomiting (14%), increased aspartate aminotransferase (12%), hyperglycemia (10%), increased alanine aminotransferase (9%), skin hyperpigmentation (8%), pneumonitis/ILD (3%), and infusion site extravasation (0.7%).
EV-201, Cohort 1
The safety of PADCEV was evaluated as a single agent in EV-201, Cohort 1 in patients (n=125) with locally advanced or mUC who had received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy
Serious adverse reactions occurred in 46% of patients treated with PADCEV. The most common serious adverse reactions (≥3%) were urinary tract infection (6%), cellulitis (5%), febrile neutropenia (4%), diarrhea (4%), sepsis (3%), acute kidney injury (3%), dyspnea (3%), and rash (3%). Fatal adverse reactions occurred in 3.2% of patients, including acute respiratory failure, aspiration pneumonia, cardiac disorder, sepsis, and pneumonitis/ILD (each 0.8%).
Adverse reactions leading to discontinuation occurred in 16% of patients; the most common adverse reaction leading to discontinuation was peripheral neuropathy (6%).
Adverse reactions leading to dose interruption occurred in 64% of patients; the most common adverse reactions leading to dose interruption were peripheral neuropathy (18%), rash (9%), and fatigue (6%).
Adverse reactions leading to dose reduction occurred in 34% of patients; the most common adverse reactions leading to dose reduction were peripheral neuropathy (12%), rash (6%), and fatigue (4%).
Table 13 summarizes the All Grades and Grades 3-4 adverse reactions reported in patients in EV-201, Cohort 1. Clinically relevant adverse reactions (<15%) include skin hyperpigmentation (14%), herpes zoster (3%), pneumonitis/ILD (2%), and infusion site extravasation (2%).
EV-201, Cohort 2
The safety of PADCEV was evaluated as a single agent in EV-201, Cohort 2 in patients with locally advanced or mUC (n=89) who received at least one dose of PADCEV 1.25 mg/kg and had prior treatment with a PD-1 or PD-L1 inhibitor and were not eligible for cisplatin-based chemotherapy. The median duration of exposure was 5.98 months (range: 0.3 to 24.6 months).
Serious adverse reactions occurred in 39% of patients treated with PADCEV. The most common serious adverse reactions (≥3%) were pneumonia, sepsis, and diarrhea (5% each). Fatal adverse reactions occurred in 8% of patients, including acute kidney injury (2.2%), metabolic acidosis, sepsis, multiorgan dysfunction, pneumonia, and pneumonitis/ILD (1.1% each).
Adverse reactions leading to discontinuation occurred in 20% of patients; the most common adverse reaction (≥2%) leading to discontinuation was peripheral neuropathy (7%).
Adverse reactions leading to dose interruption occurred in 60% of patients; the most common adverse reactions (≥3%) leading to dose interruption were peripheral neuropathy (19%), rash (9%), fatigue (8%), diarrhea (5%), increased aspartate aminotransferase (3%), and hyperglycemia (3%).
Adverse reactions leading to dose reduction occurred in 49% of patients; the most common adverse reactions (≥3%) leading to dose reduction were peripheral neuropathy (19%), rash (11%), and fatigue (7%).
Table 15 summarizes the All Grades and Grades 3-4 adverse reactions reported in patients in EV-201, Cohort 2. Clinically relevant adverse reactions (<15%) include vomiting (13%), increased aspartate aminotransferase (12%), increased lipase (11%), increased alanine aminotransferase (10%), skin hyperpigmentation (4%), pneumonitis/ILD (4%), and infusion site extravasation (1%).