Hypertrichosis-Acromegaloid Facial Appearance Syndrome Treatments
Find Hypertrichosis-Acromegaloid Facial Appearance Syndrome Treatments
Signifor
What is Signifor (Pasireotide)?
Approved To Treat
Related Clinical Trials
Summary: The researchers are doing this study to find out whether pasireotide is an effective treatment for people with prolactinoma who cannot receive dopamine agonist therapy. Another purpose of this study is to find out whether pasireotide is a safe treatment for people with prolactinoma.
Summary: Acromegaly, a chronic condition characterized by growth hormone (GH) and, in turn, insulin-like growth factor-1 (IGF-I) excess, is burdened by a series of systemic and metabolic comorbidities that strongly impair quality of life (QoL) and life expectancy. Amongst them, a specific acromegalic osteopathy has been discovered, characterized by fragility fractures associated with high bone turnover, wh...
Summary: PAMSARC is a non-commercial interventional Phase 2 clinical trial of academic research institutions, with its primary goal being to improve medical treatment of fusion driven Desmoplastic small round cell tumor (DSRCT) and Synovial sarcoma (SySa) in young adults and adolsecents with male predominance. Current management of DSRCT and SySa includes chemotherapy, radiation and aggressive cytoreductiv...
Related Latest Advances
Brand Information
- Hypocortisolism
- Hyperglycemia and Diabetes
- Bradycardia and QT prolongation
- Liver Test Elevations
- Cholelithiasis and Complications of Cholelithiasis
- Pituitary Hormone Deficiency
- Steatorrhea and Malabsorption of Dietary Fats
- Cholelithiasis resulting in complications, including cholecystitis and cholangitis, which have sometimes required cholecystectomy
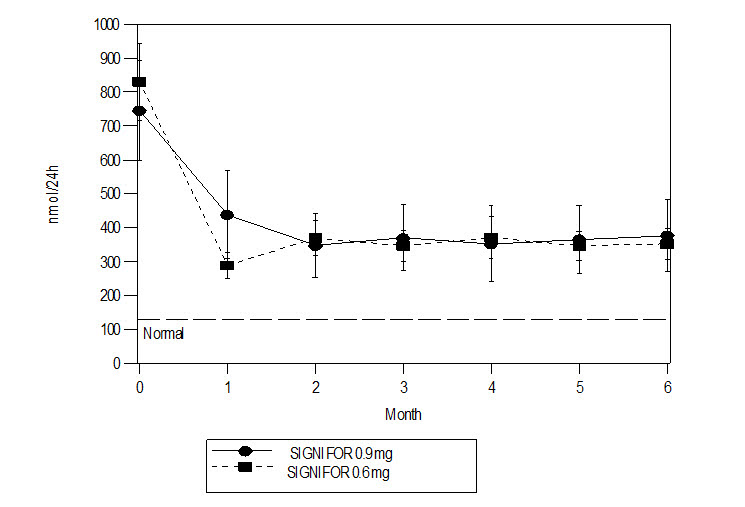
- Hypocortisolism
- Hyperglycemia and Diabetes
- Bradycardia and QT Prolongation
- Liver Test elevations
- Cholelithiasis and Complications of Cholelithiasis: Advise patients to contact their healthcare provider if they experience signs or symptoms of gallstones (cholelithiasis) or complications of gallstones (e.g., cholecystitis or cholangitis)
- Pituitary Hormone Deficiency
- Steatorrhea and Malabsorption of Dietary Fats: Advise patients to contact their healthcare provider if they experience new or worsening symptoms of steatorrhea, stool discoloration, loose stools, abdominal bloating, and weight loss
- Pregnancy: Inform female patients that treatment with SIGNIFOR may result in unintended pregnancy
- Carefully review the Medication Guide.
- Do not reuse unused portions of SIGNIFOR ampules and properly dispose of the ampules after use.
- Avoid multiple injections at or near the same site within short periods of time.


