Generic Name
Pemetrexed
Brand Names
Pemrydi Rtu, Pemfexy, Alimta, AXTLE
FDA approval date: February 04, 2004
Classification: Folate Analog Metabolic Inhibitor
Form: Injection, Solution
What is Pemrydi Rtu (Pemetrexed)?
Pemetrexed for injection is a folate analog metabolic inhibitor indicated: in combination with pembrolizumab and platinum chemotherapy, for the initial treatment of patients with metastatic non-squamous non-small cell lung cancer NSCLC, with no EGFR or ALK genomic tumor aberrations.
Approved To Treat
Top Global Experts
There are no experts for this drug
Save this treatment for later
Not sure about your diagnosis?
Tired of the same old research?
Related Clinical Trials
There is no clinical trials being done for this treatment
Related Latest Advances
There is no latest advances for this treatment
Brand Information
PEMRYDI RTU (Pemetrexed disodium)
1DOSAGE FORMS AND STRENGTHS
Injection: PEMRYDI RTU is a sterile clear colorless to pale yellow to greenish yellow solution available as follows:
- 100 mg/10 mL (10 mg/mL) single-dose vial
- 500 mg/50 mL (10 mg/mL) single-dose vial
- 1,000 mg/100 mL (10 mg/mL) single-dose vial
2CONTRAINDICATIONS
PEMRYDI RTU is contraindicated in patients with a history of severe hypersensitivity reaction to pemetrexed
3ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Myelosuppression
- Renal failure
- Bullous and exfoliative skin toxicity
- Interstitial pneumonitis
- Radiation recall
3.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
In clinical trials, the most common adverse reactions (incidence ≥ 20%) of pemetrexed, when administered as a single agent, are fatigue, nausea, and anorexia. The most common adverse reactions (incidence ≥ 20 %) of pemetrexed, when administered in combination with cisplatin are vomiting, neutropenia, anemia, stomatitis/pharyngitis, thrombocytopenia, and constipation. The most common adverse reactions (incidence ≥ 20%) of pemetrexed, when administered in combination with pembrolizumab and platinum chemotherapy, are fatigue/asthenia, nausea, constipation, diarrhea, decreased appetite, rash, vomiting, cough, dyspnea, and pyrexia.
Non-Squamous NSCLC
First-line Treatment of Metastatic Non-squamous NSCLC with Pembrolizumab and Platinum Chemotherapy
The safety of pemetrexed, in combination with pembrolizumab and investigator’s choice of platinum (either carboplatin or cisplatin), was investigated in Study KEYNOTE-189, a multicenter, double-blind, randomized (2:1), active-controlled trial in patients with previously untreated, metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations. A total of 607 patients received pemetrexed, pembrolizumab, and platinum every 3 weeks for 4 cycles followed by pemetrexed and pembrolizumab (n=405), or placebo, pemetrexed, and platinum every 3 weeks for 4 cycles followed by placebo and pemetrexed (n=202). Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible
The median duration of exposure to pemetrexed was 7.2 months (range: 1 day to 1.7 years). Seventy-two percent of patients received carboplatin. The study population characteristics were: median age of 64 years (range: 34 to 84), 49% age 65 years or older, 59% male, 94% White and 3% Asian, and 18% with history of brain metastases at baseline.
Pemetrexed was discontinued for adverse reactions in 23% of patients in the pemetrexed, pembrolizumab, and platinum arm. The most common adverse reactions resulting in discontinuation of pemetrexed in this arm were acute kidney injury (3%) and pneumonitis (2%). Adverse reactions leading to interruption of pemetrexed occurred in 49% of patients in the pemetrexed, pembrolizumab, and platinum arm. The most common adverse reactions or laboratory abnormalities leading to interruption of pemetrexed in this arm (≥ 2%) were neutropenia (12%), anemia (7%), asthenia (4%), pneumonia (4%), thrombocytopenia (4%), increased blood creatinine (3%), diarrhea (3%), and fatigue (3%).
Table 2 summarizes the adverse reactions that occurred in ≥ 20% of patients treated with pemetrexed, pembrolizumab, and platinum.
Table 2: Adverse Reactions Occurring in ≥ 20% of Patients in KEYNOTE-189
Table 3 summarizes the laboratory abnormalities that worsened from baseline in at least 20% of patients treated with pemetrexed, pembrolizumab, and platinum.
Table 3: Laboratory Abnormalities Worsened from Baseline in ≥ 20% of Patients in KEYNOTE-189
Initial Treatment in Combination with Cisplatin
The safety of pemetrexed was evaluated in Study JMDB, a randomized (1:1), open-label, multicenter trial conducted in chemotherapy-naive patients with locally advanced or metastatic NSCLC. Patients received either pemetrexed 500 mg/m
Study JMDB excluded patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS of 2 or greater), uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B
The data described below reflect exposure to pemetrexed plus cisplatin in 839 patients in Study JMDB. Median age was 61 years (range 26-83 years); 70% of patients were men; 78% were White, 16% were Asian, 2.9% were Hispanic or Latino, 2.1% were Black or African American, and <1% were other ethnicities; 36% had an ECOG PS 0. Patients received a median of 5 cycles of pemetrexed.
Table 4 provides the frequency and severity of adverse reactions that occurred in ≥ 5% of 839 patients receiving pemetrexed in combination with cisplatin in Study JMDB. Study JMDB was not designed to demonstrate a statistically significant reduction in adverse reaction rates for pemetrexed, as compared to the control arm, for any specified adverse reaction listed in Table 4.
Table 4: Adverse Reactions Occurring in ≥ 5% of Fully Vitamin-Supplemented Patients Receiving Pemetrexed in Combination with Cisplatin Chemotherapy in Study JMDB
The following additional adverse reactions were observed in patients assigned to receive pemetrexed.
Incidence 1% to < 5%
Body as a Whole — febrile neutropenia, infection, pyrexia
General Disorders — dehydration
Metabolism and Nutrition — increased AST, increased ALT
Renal —renal failure
Eye Disorder — conjunctivitis
Incidence < 1%
Cardiovascular — arrhythmia
General Disorders — chest pain
Metabolism and Nutrition — increased GGT
Neurology — motor neuropathy
Maintenance Treatment Following First-line Non-
In Study JMEN, the safety of pemetrexed was evaluated in a randomized (2:1), placebo-controlled, multicenter trial conducted in patients with non-progressive locally advanced or metastatic NSCLC following four cycles of a first-line, platinum-based chemotherapy regimen. Patients received either pemetrexed 500 mg/m
Study JMEN excluded patients with an ECOG PS of 2 or greater, uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B
The data described below reflect exposure to pemetrexed in 438 patients in Study JMEN. Median age was 61 years (range 26-83 years), 73% of patients were men; 65% were White, 31% were Asian, 2.9% were Hispanic or Latino, and < 2% were other ethnicities; 39% had an ECOG PS 0. Patients received a median of 5 cycles of pemetrexed and a relative dose intensity of pemetrexed of 96%. Approximately half the patients (48%) completed at least six, 21-day cycles and 23% completed ten or more 21-day cycles of pemetrexed.
Table 5 provides the frequency and severity of adverse reactions reported in ≥ 5% of the 438 pemetrexed-treated patients in Study JMEN.
Table 5: Adverse Reactions Occurring in ≥ 5% of Patients Receiving Pemetrexed in Study JMEN
The requirement for transfusions (9.5% versus 3.2%), primarily red blood cell transfusions, and for erythropoiesis stimulating agents (5.9% versus 1.8%) were higher in the pemetrexed arm compared to the placebo arm.
The following additional adverse reactions were observed in patients who received pemetrexed.
Incidence 1% to <5%
Dermatology/Skin — alopecia, pruritus/itching
Gastrointestinal — constipation
General Disorders — edema, fever
Hematologic — thrombocytopenia
Eye Disorder — ocular surface disease (including conjunctivitis), increased lacrimation
Incidence <1%
Cardiovascular — supraventricular arrhythmia
Dermatology/Skin — erythema multiforme
General Disorders — febrile neutropenia, allergic reaction/hypersensitivity
Neurology — motor neuropathy
Renal — renal failure
Maintenance Treatment Following First-line Pemetrexed Plus Platinum Chemotherapy
The safety of pemetrexed was evaluated in PARAMOUNT, a randomized (2:1), placebo-controlled study conducted in patients with non-squamous NSCLC with non-progressive (stable or responding disease) locally advanced or metastatic NSCLC following four cycles of pemetrexed in combination with cisplatin as first-line therapy for NSCLC. Patients were randomized to receive pemetrexed 500 mg/m
PARAMOUNT excluded patients with an ECOG PS of 2 or greater, uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B
The data described below reflect exposure to pemetrexed in 333 patients in PARAMOUNT. Median age was 61 years (range 32 to 83 years); 58% of patients were men; 94% were White, 4.8% were Asian, and < 1% were Black or African American; 36% had an ECOG PS 0. The median number of maintenance cycles was 4 for pemetrexed and placebo arms. Dose reductions for adverse reactions occurred in 3.3% of patients in the pemetrexed arm and 0.6% in the placebo arm. Dose delays for adverse reactions occurred in 22% of patients in the pemetrexed arm and 16% in the placebo arm.
Table 6 provides the frequency and severity of adverse reactions reported in ≥ 5% of the 333 pemetrexed-treated patients in PARAMOUNT.
Table 6: Adverse Reactions Occurring in ≥ 5% of Patients Receiving Pemetrexed in PARAMOUNT
The requirement for red blood cell (13% versus 4.8%) and platelet (1.5% versus 0.6%) transfusions, erythropoiesis stimulating agents (12% versus 7%), and granulocyte colony stimulating factors (6% versus 0%) were higher in the pemetrexed arm compared to the placebo arm.
The following additional Grade 3 or 4 adverse reactions were observed more frequently in the pemetrexed arm.
Incidence 1% to <5%
Blood/Bone Marrow — thrombocytopenia
General Disorders — febrile neutropenia
Incidence <1%
Cardiovascular — ventricular tachycardia, syncope
General Disorders — pain
Gastrointestinal — gastrointestinal obstruction
Neurologic — depression
Renal — renal failure
Vascular — pulmonary embolism
Treatment of Recurrent Disease After Prior Chemotherapy
The safety of pemetrexed was evaluated in Study JMEI, a randomized (1:1), open-label, active-controlled trial conducted in patients who had progressed following platinum-based chemotherapy. Patients received pemetrexed 500 mg/m
Study JMEI excluded patients with an ECOG PS of 3 or greater, uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to discontinue aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B
The data described below reflect exposure to pemetrexed in 265 patients in Study JMEI. Median age was 58 years (range 22 to 87 years); 73% of patients were men; 70% were White, 24% were Asian, 2.6% were Black or African American, 1.8% were Hispanic or Latino, and < 2% were other ethnicities; 19% had an ECOG PS 0.
Table 7 provides the frequency and severity of adverse reactions reported in ≥ 5% of the 265 pemetrexed-treated patients in Study JMEI. Study JMEI is not designed to demonstrate a statistically significant reduction in adverse reaction rates for pemetrexed, as compared to the control arm, for any specified adverse reaction listed in the Table 7 below.
Table 7: Adverse Reactions Occurring in ≥ 5% of Fully Supplemented Patients Receiving Pemetrexed in Study JMEI
The following additional adverse reactions were observed in patients assigned to receive pemetrexed.
Incidence 1% to <5%
Body as a Whole — abdominal pain, allergic reaction/hypersensitivity, febrile neutropenia, infection
Dermatology/Skin — erythema multiforme
Neurology — motor neuropathy, sensory neuropathy
Incidence <1%
Cardiovascular — supraventricular arrhythmias
Renal — renal failure
Mesothelioma
The safety of pemetrexed was evaluated in Study JMCH, a randomized (1:1), single-blind study conducted in patients with MPM who had received no prior chemotherapy for MPM. Patients received pemetrexed 500 mg/m
Study JMCH excluded patients with Karnofsky Performance Scale (KPS) of less than 70, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs were also excluded from the study.
The data described below reflect exposure to pemetrexed in 168 patients that were fully supplemented with folic acid and vitamin B
Table 8 provides the frequency and severity of adverse reactions ≥ 5% in the subgroup of pemetrexed-treated patients who were fully vitamin supplemented in Study JMCH. Study JMCH was not designed to demonstrate a statistically significant reduction in adverse reaction rates for pemetrexed, as compared to the control arm, for any specified adverse reaction listed in the table below.
Table 8: Adverse Reactions Occurring in ≥ 5% of Fully Supplemented Subgroup of Patients Receiving Pemetrexed/Cisplatin in Study JMCH
The following additional adverse reactions were observed in patients receiving pemetrexed plus cisplatin:
Incidence 1% to <5%
Body as a Whole — febrile neutropenia, infection, pyrexia
Dermatology/Skin — urticaria
General Disorders — chest pain
Metabolism and Nutrition — increased AST, increased ALT, increased GGT
Renal — renal failure
Incidence <1%
Cardiovascular — arrhythmia
Neurology — motor neuropathy
Exploratory Subgroup Analyses based on Vitamin Supplementation
Table 9 provides the results of exploratory analyses of the frequency and severity of NCI CTCAE Grade 3 or 4 adverse reactions reported in more pemetrexed-treated patients who did not receive vitamin supplementation (never supplemented) as compared with those who received vitamin supplementation with daily folic acid and vitamin B
Table 9: Exploratory Subgroup Analysis of Selected Grade 3/4 Adverse Reactions Occurring in Patients Receiving Pemetrexed in Combination with Cisplatin with or without Full Vitamin Supplementation in Study JMCH
The following adverse reactions occurred more frequently in patients who were fully vitamin supplemented than in patients who were never supplemented:
- hypertension (11% versus 3%),
- chest pain (8% versus 6%),
- thrombosis/embolism (6% versus 3%).
Additional Experience Across Clinical Trials
Sepsis, with or without neutropenia, including fatal cases: 1%
Severe esophagitis, resulting in hospitalization: <1%
3.2Postmarketing Experience
The following adverse reactions have been identified during post-approval use of pemetrexed. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System — immune-mediated hemolytic anemia
Gastrointestinal — colitis, pancreatitis
General Disorders and Administration Site Conditions — edema
Injury, poisoning, and procedural complications — radiation recall
Respiratory — interstitial pneumonitis
Skin — Serious and fatal bullous skin conditions, Stevens-Johnson syndrome, and toxic epidermal necrolysis
4DRUG INTERACTIONS
Effects of Ibuprofen on Pemetrexed
Ibuprofen increases exposure (AUC) of pemetrexed
- Avoid administration of ibuprofen for 2 days before, the day of, and 2 days following administration of PEMRYDI RTU
- Monitor patients more frequently for myelosuppression, renal, and gastrointestinal toxicity, if concomitant administration of ibuprofen cannot be avoided.
5OVERDOSAGE
No drugs are approved for the treatment of pemetrexed overdose. Based on animal studies, administration of leucovorin may mitigate the toxicities of pemetrexed overdosage. It is not known whether pemetrexed is dialyzable.
6DESCRIPTION
Pemetrexed is a folate analog metabolic inhibitor. The drug substance, Pemetrexed Disodium Hemipentahydrate, has the chemical name disodium N-[4-[2-(2-amino-4,7-dihydro-4-oxo-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl] benzoyl]-L-glutamic acid hemipentahydrate, with a molecular formula of C
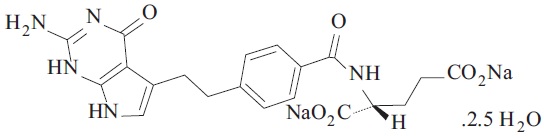
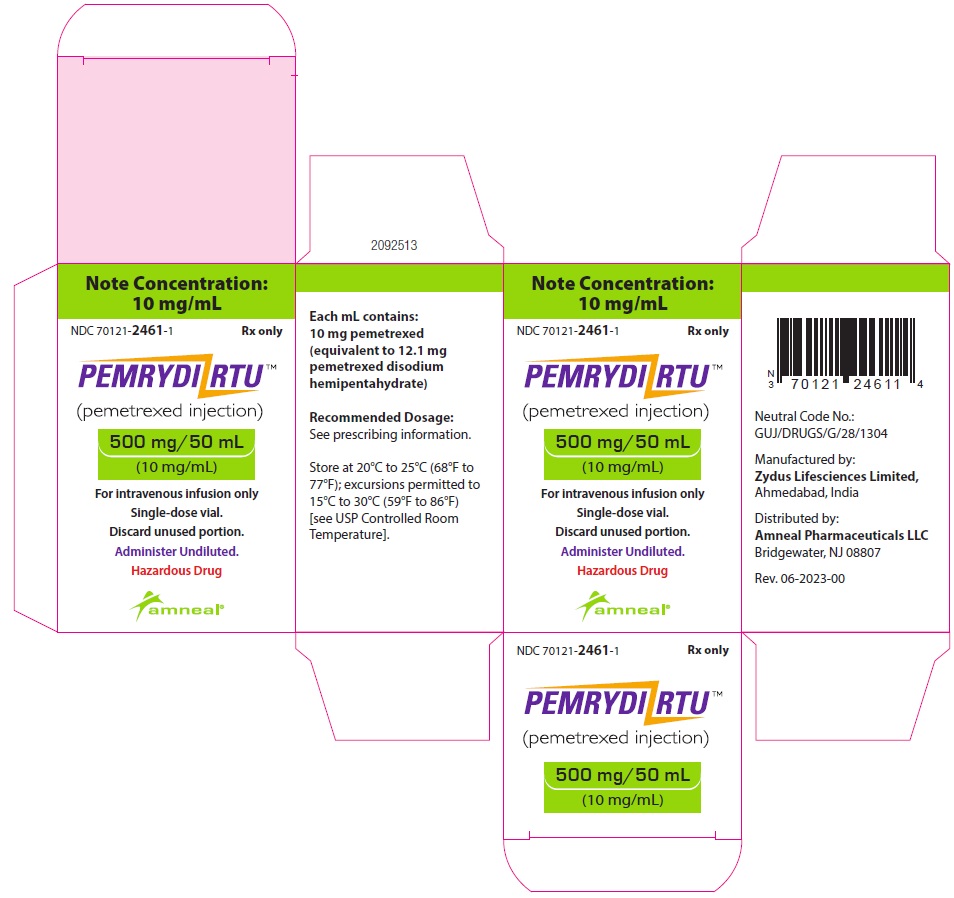
PEMRYDI RTU is a sterile clear, colorless to pale yellow to green-yellow ready-to-use solution in single-dose vials. Each milliliter of solution contains 10 mg of pemetrexed (equivalent to 12.1 mg pemetrexed disodium hemipentahydrate), 10 mg of mannitol, 9 mg of sodium chloride, 1 mg of L-cysteine hydrochloride, sodium hydroxide and/or hydrochloric acid to adjust pH and water for injection.
7REFERENCES
1. “OSHA Hazardous Drugs” OSHA
8HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
PEMRYDI RTU (pemetrexed injection) is supplied as a sterile clear, colorless to pale yellow to green-yellow ready-to-use solution packaged in a USP type-I glass vial with rubber stopper and aluminium flip-off cap.
It is available as follows:
100 mg/10 mL (10 mg/mL):
1 Single-dose Vial in a Carton: NDC 70121-2453-1
500 mg/50 mL (10 mg/mL):
1 Single-dose Vial in a Carton: NDC 70121-2461-1
1,000 mg/100 mL (10 mg/mL):
1 Single-dose Vial in a Carton: NDC 70121-2462-1
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
PEMRYDI RTU is a hazardous drug. Follow applicable special handling and disposal procedures.
9PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Premedication and Concomitant Medication: Instruct patients to take folic acid as directed and to keep appointments for vitamin B12 injections to reduce the risk of treatment-related toxicity. Instruct patients of the requirement to take corticosteroids to reduce the risks of treatment-related toxicity [see and Warnings and Precautions (5.1)].
Myelosuppression: Inform patients of the risk of low blood cell counts and instruct them to immediately contact their physician for signs of infection, fever, bleeding, or symptoms of anemia [see .
Renal Failure: Inform patients of the risks of renal failure, which may be exacerbated in patients with dehydration arising from severe vomiting or diarrhea. Instruct patients to immediately contact their healthcare provider for a decrease in urine output [see .
Bullous and Exfoliative Skin Disorders: Inform patients of the risks of severe and exfoliative skin disorders. Instruct patients to immediately contact their healthcare provider for development of bullous lesions or exfoliation in the skin or mucous membranes [see .
Interstitial Pneumonitis: Inform patients of the risks of pneumonitis. Instruct patients to immediately contact their healthcare provider for development of dyspnea or persistent cough [see .
Radiation Recall: Inform patients who have received prior radiation of the risks of radiation recall. Instruct patients to immediately contact their healthcare provider for development of inflammation or blisters in an area that was previously irradiated [see .
Increased Risk of Toxicity with Ibuprofen in Patients with Renal Impairment: Advise patients with mild to moderate renal impairment of the risks associated with concomitant ibuprofen use and instruct them to avoid use of all ibuprofen containing products for 2 days before, the day of, and 2 days following administration of PEMRYDI RTU [see and Drug Interactions (7)].
Embryo-Fetal Toxicity: Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus [see and Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 6 months after the last dose. Advise females to inform their prescriber of a known or suspected pregnancy. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 3 months after the last dose [see and Use in Specific Populations (8.3)].
Lactation: Advise women not to breastfeed during treatment with PEMRYDI RTU and for 1 week after the last dose [see .
PEMRYDI RTU
Manufactured by:
Zydus Lifesciences Limited
Ahmedabad, India.
Distributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 07-2025-03
10PATIENT INFORMATION
This Patient Information has been approved by the U.S. Food and Drug Administration.
11PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC 70121-2453-1
PEMRYDI RTU (pemetrexed injection)
100 mg/10 mL (10 mg/mL)
For Intravenous infusion only
Rx only
Amneal Pharmaceuticals LLC

NDC 70121-2461-1
PEMRYDI RTU (pemetrexed injection)
500 mg/50 mL (10 mg/mL)
For Intravenous infusion only
Rx only
Amneal Pharmaceuticals LLC
NDC 70121-2462-1
PEMRYDI RTU (pemetrexed injection)
1,000 mg/100 mL (10 mg/mL)
For Intravenous infusion only
Rx only
Amneal Pharmaceuticals LLC