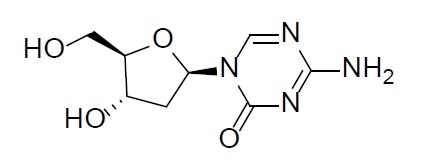
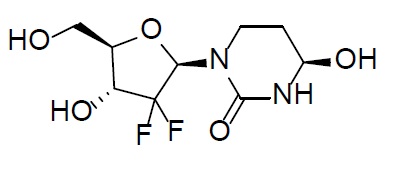
Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.
INQOVI Monotherapy for MDS or CMML
The safety of INQOVI was evaluated in a pooled safety population that includes patients enrolled in Study ASTX727-01-B and Study ASTX727-02
Patients were randomized to receive INQOVI (35 mg decitabine and 100 mg cedazuridine) orally once daily on Days 1 through 5 in Cycle 1 and decitabine 20 mg/m
Serious adverse reactions occurred in 68% of patients who received INQOVI. Serious adverse reactions in > 5% of patients included febrile neutropenia (30%), pneumonia (14%), and sepsis (13%). Fatal adverse reactions occurred in 6% of patients. These included sepsis (1%), septic shock (1%), pneumonia (1%), respiratory failure (1%), and one case each of cerebral hemorrhage and sudden death.
Permanent discontinuation due to an adverse reaction occurred in 5% of patients who received INQOVI. The most frequent adverse reactions resulting in permanent discontinuation were febrile neutropenia (1%) and pneumonia (1%).
Dose interruptions due to an adverse reaction occurred in 41% of patients who received INQOVI. Adverse reactions requiring dosage interruptions in > 5% of patients who received INQOVI included neutropenia (18%), febrile neutropenia (8%), thrombocytopenia (6%), and anemia (5%).
Dose reductions due to an adverse reaction occurred in 19% of patients who received INQOVI. Adverse reactions requiring dosage reductions in >2% of patients who received INQOVI included neutropenia (12%), anemia (3%), and thrombocytopenia (3%).
The most common adverse reactions (≥20%) were fatigue, constipation, hemorrhage, myalgia, mucositis, arthralgia, nausea, dyspnea, diarrhea, rash, dizziness, febrile neutropenia, edema, headache, cough, decreased appetite, upper respiratory tract infection, pneumonia, and transaminase increased. The most common Grade 3 or 4 laboratory abnormalities (≥ 50%) were leukocytes decreased, platelet count decreased, neutrophil count decreased, and hemoglobin decreased.
Table 3 summarizes the adverse reactions in the pooled safety population. Clinically relevant adverse reactions in < 10% of patients who received INQOVI included:
- Acute febrile neutrophilic dermatosis (Sweet’s syndrome) (1%)
- Tumor lysis syndrome (0.5%)
INQOVI in Combination with Venetoclax for AML
The safety of INQOVI in combination with venetoclax (INQOVI+VEN) in adult patients 75 years of age and older or those who have comorbidities precluding the use of intensive induction chemotherapy was evaluated in Study ASTX727-07 (N=159)
Patients received INQOVI (35 mg decitabine and 100 mg cedazuridine) orally once daily on Days 1 through 5 of the first 28-day cycle in combination with venetoclax given as a ramp up on Day 1 (100 mg) and Day 2 (200 mg), followed by venetoclax 400 mg daily from Day 3 onward.
Among the patients who received INQOVI+VEN, the median duration of exposure was 5.5 months (range 0.2-28 months): 47% of patients were exposed to INQOVI for 6 months or longer and 20% of patients were exposed to INQOVI for greater than 1 year.
Serious adverse reactions occurred in 82% of patients who received INQOVI+VEN. Serious adverse reactions in > 5% of patients included febrile neutropenia (31%), sepsis (22%), pneumonia (15%), infection (bacterial/viral) (10%), hemorrhage (9%), and dyspnea (6%). Fatal adverse reactions occurred in 8% of patients who received INQOVI+VEN. These included sepsis (5%), dyspnea (2%), myocardial infarction (1%), hemolytic anemia (1%), and tumor lysis syndrome (1%).
Permanent discontinuation of INQOVI due to an adverse reaction occurred in 9% of patients who received INQOVI+VEN. The most frequent adverse reaction resulting in permanent discontinuation in more than 1 patient was hemorrhage (1%).
Dosage interruptions of INQOVI due to an adverse reaction occurred in 55% of patients who received INQOVI+VEN. Adverse reactions requiring dosage interruptions in ≥ 5% of patients who received INQOVI+VEN included neutropenia (40%), febrile neutropenia (11%), infection (bacterial/viral) (8%), and thrombocytopenia (8%).
Dose reductions of INQOVI due to an adverse reaction occurred in 6% of patients who received INQOVI+VEN. The most common adverse reactions requiring dosage reductions of INQOVI were neutropenia (4%), thrombocytopenia (1%), and infection (1%).
The most common adverse reactions (≥ 20%) were neutropenia, febrile neutropenia, thrombocytopenia, hemorrhage, anemia, infection (bacterial/viral), diarrhea, fatigue, mucositis, constipation, arthralgia, dyspnea, decreased appetite, edema, nausea, white blood cell count decreased, sepsis, pneumonia, rash, transaminitis, myalgia, arrhythmia, and abdominal pain. The most common Grade 3 or 4 laboratory abnormalities (≥ 50%) were decreased leukocytes, decreased lymphocytes, decreased platelets, and decreased hemoglobin.
Table 5 summarizes the adverse reactions in ASTX727-07. Clinically relevant adverse reactions in < 10% of patients who received INQOVI+VEN included:
- Acute febrile neutrophilic dermatosis (Sweet’s syndrome) (1%)
- Tumor lysis syndrome (2%)
Table 6 summarizes the laboratory abnormalities identified in the combined AML safety population.