Brand Name
Movantik
Generic Name
Naloxegol
View Brand Information FDA approval date: October 01, 2020
Classification: Opioid Antagonist
Form: Tablet
What is Movantik (Naloxegol)?
MOVANTIK ® is indicated for the treatment of opioid-induced constipation in adult patients with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent opioid dosage escalation. MOVANTIK is an opioid antagonist indicated for the treatment of opioid-induced constipation in adult patients with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent opioid dosage escalation.
Approved To Treat




Save this treatment for later
Not sure about your diagnosis?
Related Clinical Trials
Risk of Major Adverse Cardiovascular Events Among Users of Naldemedine Compared With Other Medications Used for Opioid Induced Constipation in Adult Patients With Chronic Non-Cancer Pain in a Healthcare Claims Database
Summary: The research objective is to characterize the risk of a major adverse cardiovascular event (MACE) among new users of naldemedine versus new users of lubiprostone and new users of naloxegol as comparator opioid induced constipation (OIC) medications.
The NIPA Study: a Randomized Double-blind Control Clinical Trial Naloxegol Administration to Prevent Opioids Induced Gastrointestinal Motility Disturbance in Brain Injured PAtients
Summary: Impaired gastrointestinal transit (IGT) especially constipation, is common among patients under mechanical ventilation, occurring in up to 80 % of the patients during the first week, and has been associated with worse outcome in intensive care unit (ICU). Although IGT in critically ill patients is multifactorial and some components are due to complex disease, there is increasing evidence that exog...
Related Latest Advances
Brand Information
MOVANTIK (naloxegol oxalate)
1INDICATIONS AND USAGE
MOVANTIK
2DOSAGE FORMS AND STRENGTHS
MOVANTIK (naloxegol) is available in two strengths:
• Tablets: 12.5 mg supplied as mauve, oval, biconvex, film-coated, intagliated with “nGL” on one side and “12.5” on the other side.
3CONTRAINDICATIONS
MOVANTIK is contraindicated in:
- Patients with known or suspected gastrointestinal obstruction and patients at risk of recurrent obstruction, due to the potential for gastrointestinal perforation
- Patients concomitantly using strong CYP3A4 inhibitors (e.g., clarithromycin, ketoconazole) because these medications can significantly increase exposure to naloxegol which may precipitate opioid withdrawal symptoms such as hyperhidrosis, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning
- Patients who have had a known serious or severe hypersensitivity reaction to MOVANTIK or any of its excipients
4ADVERSE REACTIONS
Serious and important adverse reactions described elsewhere in labeling include:
- Opioid withdrawal
- Severe abdominal pain and/or diarrhea
- Gastrointestinal perforation
4.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to MOVANTIK in 1497 patients in clinical trials, including 537 patients exposed for greater than six months, and 320 patients exposed for 12 months.
The safety data described in Table 1 are derived from two double-blind, placebo-controlled trials (Studies 1 and 2) in patients with OIC and non-cancer related pain
Table 1lists adverse reactions in pooled Studies 1 and 2 occurring in ≥3% of patients receiving MOVANTIK 12.5 mg or 25 mg and at an incidence greater than placebo.
Opioid Withdrawal
Possible opioid withdrawal, defined as at least three adverse reactions potentially related to opioid withdrawal that occurred on the same day and were not all related to the gastrointestinal system, occurred in less than 1% (1/444) of placebo subjects, 1% (5/441) receiving MOVANTIK 12.5 mg, and 3% (14/446) receiving MOVANTIK 25 mg in Studies 1 and 2 regardless of maintenance opioid treatment. Symptoms included but were not limited to hyperhidrosis, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning. Patients receiving methadone as therapy for their pain condition were observed in Studies 1 and 2 to have a higher frequency of gastrointestinal adverse reactions than patients receiving other opioids [39% (7/18) vs. 26% (110/423) in the 12.5 mg group; 75% (24/32) vs. 34% (142/414) in the 25 mg group].
4.2Postmarketing Experience
The following adverse reactions have been identified during post-approval use of MOVANTIK. Because reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Hypersensitivity reactions: angioedema, rash, and urticaria.
Gastrointestinal disorders: Gastrointestinal perforation [see .
5OVERDOSAGE
In a clinical study of patients with OIC a daily dose of 50 mg (twice the recommended dosage), administered over 4 weeks, was associated with an increased incidence of GI adverse reactions, such as abdominal pain, diarrhea, and nausea. These adverse reactions frequently occurred within 1-2 days after dosing.
No antidote is known for naloxegol. Dialysis was noted to be ineffective as a means of elimination in a clinical study in patients with renal failure.
If a patient on opioid therapy receives an overdose of naloxegol, the patient should be monitored closely for potential evidence of opioid withdrawal symptoms such as chills, rhinorrhea, diaphoresis, or reversal of central analgesic effect. Base treatment on the degree of opioid withdrawal symptoms, including changes in blood pressure and heart rate, and on the need for analgesia.
6DESCRIPTION
MOVANTIK (naloxegol), an opioid antagonist, contains naloxegol oxalate as the active ingredient. (Naloxegol is a PEGylated derivative of naloxone.)
The chemical name for naloxegol oxalate is: (5α,6α)-17-allyl-6-(2,5,8,11,14,17,20-heptaoxadocosan-22-yloxy)-4,5-epoxymorphinan-3,14-diol oxalate. The structural formula is:
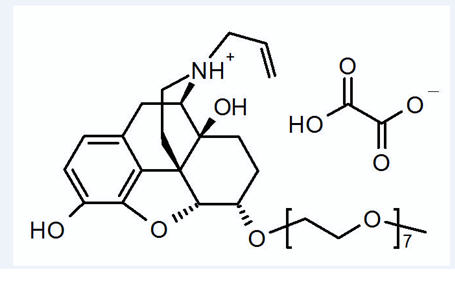
The empirical formula for naloxegol oxalate is C
Naloxegol oxalate is a white to off-white powder, with high aqueous solubility across the physiologic pH range.
MOVANTIK (naloxegol) tablets for oral use contain 14.2 mg and 28.5 mg of naloxegol oxalate, respectively, equivalent to 12.5 mg and 25 mg of naloxegol.
Excipients in tablet core are: mannitol, cellulose microcrystalline, croscarmellose sodium, magnesium stearate, and propyl gallate.
Excipients in tablet coat are: hypromellose, titanium dioxide, polyethylene glycol, iron oxide red, and iron oxide black.
7CLINICAL STUDIES
The safety and efficacy of MOVANTIK were evaluated in two replicate, randomized, double-blind placebo-controlled trials (Study 1 and Study 2) in patients with opioid-induced constipation (OIC) and non-cancer related pain.
Patients receiving an opioid morphine equivalent daily dose of between 30 mg and 1,000 mg for at least four weeks before enrollment and self-reported OIC were eligible to participate. OIC was confirmed through a two-week run-in period and was defined as <3 spontaneous bowel movements (SBMs) per week on average with at least 25% of the SBMs associated with one or more of the following conditions: (1) straining, (2) hard or lumpy stools; and (3) having a sensation of incomplete evacuation. An SBM was defined as a bowel movement (BM) without rescue laxative taken within the past 24 hours. Patients with 0 BMs over the two-week run-in period or patients with an uneven distribution of SBMs across the two-week run-in period (0 SBMs in one week with ≥4 SBMs in the other week) were excluded. Throughout the studies (including the two-week run-in period), patients were prohibited from using laxatives other than bisacodyl rescue laxative (if they had not had a BM for 72 hours) and one-time use of an enema (if after 3 doses of bisacodyl, they still did not have a BM).
Patients suspected of having clinically important disruptions to the blood-brain barrier were not enrolled in these studies.
A total of 652 patients in Study 1 and 700 patients in Study 2 were randomized in a 1:1:1 ratio to receive 12.5 mg or 25 mg of MOVANTIK or placebo once daily for 12 weeks.
The mean age of the subjects in these two studies was 52 years, 10% and 13% were 65 years of age or older, 61% and 63% were women, and 78% and 80% were White in Studies 1 and 2, respectively.
Back pain was the most common reason for pain (56% and 57%); arthritis (10% and 10%) and joint pain (3% and 5%) were other prominent reasons in Studies 1 and 2, respectively. Prior to enrollment, patients had been using their current opioid for an average of 3.6 and 3.7 years. The patients who participated in Studies 1 and 2 were taking a wide range of opioids. The mean baseline opioid morphine equivalent daily dosage was 140 mg and 136 mg per day.
Use of one or more laxatives on at least one occasion within the two weeks prior to enrollment was reported by 71% of patients in both Studies 1 and 2.
The primary endpoint was response defined as: ≥3 SBMs per week and a change from baseline of ≥1 SBM per week for at least 9 out of the 12 study weeks and 3 out of the last 4 weeks.
There was a statistically significant difference for the 25 mg MOVANTIK treatment group versus placebo for the primary endpoint in Study 1 and Study 2 (see
One secondary endpoint in Study 1 and Study 2 was response in laxative users with OIC symptoms. This subgroup comprised 55% and 53% of total patients in these two studies, respectively. These patients (identified using an investigator-administered questionnaire), prior to enrollment, had reported using laxative(s) at least 4 out of the past 14 days with at least one of the following OIC symptoms of moderate, severe, or very severe intensity: incomplete bowel movements, hard stool, straining, or sensation of needing to pass a bowel movement but unable to do so. In this subgroup, in Studies 1 and 2, 42% and 50% reported using laxatives on a daily basis. The most frequently reported laxatives used on a daily basis were stool softeners (18% and 24%), stimulants (16% and 18%), and polyethylene glycol (6% and 5%). Use of two laxative classes was reported in 31% and 27% anytime during the 14 days prior to enrollment. The most commonly reported combination was stimulants and stool softeners (10% and 8%). In Study 1, a statistically significantly higher percentage of patients in this subgroup responded with MOVANTIK 12.5 mg compared to placebo (43% vs. 29%; p=0.03) and with MOVANTIK 25 mg compared to placebo (49% vs. 29%; p=0.002). In Study 2, a statistically significantly higher percentage of patients in this subgroup responded with MOVANTIK 25 mg compared to placebo (47% vs. 31%; p=0.01). This secondary endpoint was not tested for MOVANTIK 12.5 mg versus placebo in Study 2 because the primary endpoint was not statistically significant.
Another secondary endpoint was time to first post-dose SBM. The time to first post-dose SBM was significantly shorter with MOVANTIK 25 mg compared to placebo in both Study 1 (p <0.001) and Study 2 (p <0.001), and for MOVANTIK 12.5 mg as compared to placebo in Study 1 (p <0.001). For Study 1, the median times to first post-dose SBM were 6, 20, and 36 hours with MOVANTIK 25 mg, MOVANTIK 12.5 mg, and placebo, respectively. For Study 2, the median times to first post-dose SBM were 12 and 37 hours with MOVANTIK 25 mg and placebo, respectively. These analyses do not include the results for MOVANTIK 12.5 mg versus placebo in Study 2 because the primary endpoint was not statistically significant. In the two studies, 61-70% and 58% of patients receiving MOVANTIK 25 mg and MOVANTIK 12.5 mg, respectively, had an SBM within 24 hours of the first dose.
A third secondary endpoint was an evaluation of change from baseline between the treatment groups for mean number of days per week with at least 1 SBM but no more than 3 SBMs. There was a significant difference in number of days per week with 1 to 3 SBMs per day on average over 12 weeks between MOVANTIK 25 mg (Study 1 and Study 2) and MOVANTIK 12.5 mg (Study 1) and placebo.
8HOW SUPPLIED/STORAGE AND HANDLING
MOVANTIK (naloxegol) tablets are supplied as:
- NDC 82625-8801-1: 12.5 mg, bottle of 30 tablets
- NDC 82625-8801-2: 12.5 mg, bottle of 90 tablets
- NDC 82625-8801-3: 12.5 mg, unit dose blister carton of 100 tablets (for HUD only)
- NDC 82625-8802-1: 25 mg, bottle of 30 tablets
- NDC 82625-8802-2: 25 mg, bottle of 90 tablets
- NDC 82625-8802-3: 25 mg, unit dose blister carton of 100 tablets (for HUD only)
Storage
Store MOVANTIK at 20-25°C (68-77°F). Excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature].
9PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration
Advise patients to:
- Discontinue all maintenance laxative therapy prior to initiation of MOVANTIK. Laxative(s) can be used as needed if there is a suboptimal response to MOVANTIK after three days.
- Take MOVANTIK on an empty stomach at least 1 hour prior to the first meal of the day or 2 hours after the meal.
- Discontinue MOVANTIK if treatment with the opioid pain medication is also discontinued.
- Avoid consumption of grapefruit or grapefruit juice during treatment with MOVANTIK.
- Inform their healthcare provider if their opioid pain medication is discontinued.
- Inform their healthcare provider if they are unable to tolerate MOVANTIK, so a dosage adjustment can be considered.
- If patients are unable to swallow the MOVANTIK tablet whole, the tablet can be crushed to a powder, mixed with water and administered orally or via a nasogastric (NG) tube, as described in the Medication Guide.
Drug Interactions
Advise patients to tell their healthcare provider when they start or stop taking any concomitant medications. Strong CYP3A4 inhibitors (e.g., clarithromycin, ketoconazole) are contraindicated with MOVANTIK, and other CYP3A4 enzyme modulating drugs can alter MOVANTIK exposure
Opioid Withdrawal
Advise patients that clusters of symptoms consistent with opioid withdrawal may occur while taking MOVANTIK, including sweating, chills, diarrhea, abdominal pain, anxiety, irritability, and yawning. Inform patients taking methadone as therapy for their pain condition that they may be more likely to have gastrointestinal adverse reactions such as abdominal pain and diarrhea that may be related to opioid withdrawal, than patients receiving other opioids
Severe Abdominal Pain and/or Diarrhea
Advise patients that symptoms may occur after starting treatment. The patient should discontinue MOVANTIK and contact their healthcare provider if they develop severe abdominal pain and/or diarrhea
Gastrointestinal Perforation
Advise patients to discontinue MOVANTIK and promptly seek medical attention if they develop unusually severe, persistent or worsening abdominal pain
Pregnancy
Advise females of reproductive potential, who become pregnant or are planning to become pregnant that the use of MOVANTIK during pregnancy may precipitate opioid withdrawal in the pregnant women and the fetus
Lactation
Advise females that breastfeeding is not recommended during treatment with MOVANTIK
MOVANTIK is a registered trademark of the AstraZeneca Group of companies and used under license by Valinor Pharma, LLC.
Distributed by: Valinor Pharma, LLC, Chicago, IL 60606
© 2023 Valinor Pharma, LLC
10MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 03/2023
11PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 12.5 mg
NDC 82625-8801-1
movantik
(naloxegol) Tablets
12.5 mg*
*Each tablet contains 14.2 mg
Dispense the accompanying
Rx only
Valinor
12PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 25 mg
NDC 82625-8802-1
movantik
(naloxegol) Tablets
25 mg*
*Each tablet contains 28.5 mg
Dispense the accompanying
Rx only
Valinor