Brand Name
Saphnelo
Generic Name
Anifrolumab-fnia
View Brand Information FDA approval date: July 30, 2021
Classification: Type I Interferon Receptor Antagonist
Form: Injection
What is Saphnelo (Anifrolumab-fnia)?
SAPHNELO is indicated for the treatment of adult patients with moderate to severe systemic lupus erythematosus , who are receiving standard therapy. Limitations of Use The efficacy of SAPHNELO has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus. Use of SAPHNELO is not recommended in these situations. SAPHNELO is a type I interferon receptor antagonist indicated for the treatment of adult patients with moderate to severe systemic lupus erythematosus , who are receiving standard therapy. Limitations of Use: The efficacy of SAPHNELO has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus. Use of SAPHNELO is not recommended in these situations.
Approved To Treat
Top Global Experts
Save this treatment for later
Not sure about your diagnosis?
Related Clinical Trials
There is no clinical trials being done for this treatment
Related Latest Advances
Brand Information
SAPHNELO (Anifrolumab-fnia)
1INDICATIONS AND USAGE
SAPHNELO (anifrolumab-fnia) is indicated for the treatment of adult patients with moderate to severe systemic lupus erythematosus (SLE), who are receiving standard therapy
Limitations of Use
The efficacy of SAPHNELO has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus. Use of SAPHNELO is not recommended in these situations.
2DOSAGE FORMS AND STRENGTHS
Injection: 300 mg/2 mL (150 mg/mL) as a clear to opalescent, colorless to slightly yellow, solution in a single-dose vial.
3CONTRAINDICATIONS
SAPHNELO is contraindicated in patients with a history of anaphylaxis with anifrolumab-fnia
4ADVERSE REACTIONS
The following clinically significant adverse reactions are also discussed elsewhere in the labeling:
- Serious Infections
- Hypersensitivity Reactions Including Anaphylaxis
- Malignancy
4.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of SAPHNELO was assessed through 52 weeks in patients with moderate to severe SLE who received anifrolumab-fnia 300 mg by intravenous infusion every 4 weeks (N=459), compared to placebo (N=466) in controlled clinical trials (Trials 1, 2 and 3) [see
In the controlled-clinical trials, adverse reactions, irrespective of causality, were reported in 87% of patients receiving SAPHNELO and 79% of patients receiving placebo.
Adverse reactions that occurred at greater than or equal to 2% incidence are shown in Table 1.
Long-term Safety
Patients who completed Trials 2 and 3 (Phase III feeder trials) were eligible to continue on treatment in a randomized, double-blind, placebo-controlled long-term extension (LTE) study, for an additional 3 years. The long-term safety of SAPHNELO was assessed in 257 patients who received anifrolumab-fnia 300 mg and 112 patients who received placebo in both a feeder trial and the LTE. Of these, 177 patients who received SAPHNELO (68.9%) and 52 patients who received placebo (46.4%) completed a total of 4 years on treatment. The overall long-term safety profile of SAPHNELO was consistent with Trials 1, 2 and 3.
Specific Adverse Reactions
Infections
In the 52‑week controlled-clinical trials, infections were reported in a greater proportion of patients while on treatment with SAPHNELO compared to placebo (69.7% [320/459] versus 55.4% [258/466]), corresponding to exposure-adjusted incidence rates (EAIR) of 141.8 and 99.9 per 100 patient years (PY), respectively.
Serious Infections
In the 52‑week controlled-clinical trials, the incidence of serious infections while on treatment was 4.8% (22/459) in patients treated with SAPHNELO compared with 5.6% (26/466) in patients receiving placebo, corresponding to EAIR of 5.4 and 6.6 per 100 PY, respectively. The most frequent serious infection was pneumonia.
In the 52‑week controlled-clinical trials, fatal infections occurred in 0.4% of patients receiving SAPHNELO and 0.2% of the patients receiving placebo.
During the LTE study, the most common serious infections were COVID-19 and pneumonia.
Herpes Zoster
In the 52‑week controlled-clinical trials, the incidence of herpes zoster in patients while on treatment with SAPHNELO was 6.1% (28/459) and 1.3% (6/466) in patients on placebo, corresponding to EAIRs of 6.9 and 1.5 per 100 PY, respectively. Cases with multidermatomal involvement and disseminated presentation have been reported. Of the 28 SAPHNELO-treated patients with herpes zoster, 2 experienced disseminated disease requiring hospitalization compared to none among patients who received placebo.
Hypersensitivity Reactions Including Anaphylaxis
During the SLE development program, there was one report of an anaphylactic reaction in a patient who received 150 mg anifrolumab-fnia, and 4 reports of angioedema after 300 mg. In general, the hypersensitivity reactions were predominantly mild or moderate in intensity and did not lead to discontinuation of SAPHNELO.
In the 52‑week controlled-clinical trials, hypersensitivity reactions occurred in 2.8% (13/459) of patients while on treatment with SAPHNELO and 0.6% (3/466) of patients on placebo, corresponding to EAIR of 3.2 and 0.7 per 100 PY, respectively. Serious hypersensitivity reactions were reported for 0.6% (3/459) of patients receiving SAPHNELO, including angioedema (n=2).
Infusion-related Reactions
Infusion-related reactions were mild to moderate in intensity; the most common symptoms were headache, nausea, vomiting, fatigue, and dizziness.
In the 52‑week controlled-clinical trials, the incidence of infusion-related reactions while on treatment was 9.4% (43/459) in patients while on treatment with SAPHNELO and 7.1% (33/466) in patients on placebo, corresponding to EAIRs of 11.1 and 8.7 per 100 PY, respectively.
Malignancies
In 52‑week controlled-clinical trials, malignancies (excluding non-melanoma skin cancers) were observed in 0.7% (3/459) and 0.6% (3/466) of patients receiving SAPHNELO and placebo, corresponding to EAIR of 0.7 and 0.7 per 100 PY, respectively. Malignant neoplasm (including non-melanoma skin cancers) was reported for 1.3% (6/459) patients receiving SAPHNELO, compared to 0.6% (3/466) patients receiving placebo (EAIR: 1.3 and 0.7 per 100 PY, respectively). The malignancies that were reported in more than one patient treated with SAPHNELO included breast cancer and squamous cell carcinoma.
4.2Postmarketing Experience
The following adverse reaction has been identified during post-approval use of SAPHNELO. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Arthralgia
5DRUG INTERACTIONS
No formal drug interaction studies have been conducted.
6DESCRIPTION
Anifrolumab-fnia is a type I interferon (IFN) receptor antagonist, immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that is produced in mouse myeloma cells (NS0) by recombinant DNA technology. The molecular weight is approximately 148 kDa.
SAPHNELO (anifrolumab-fnia) injection is a sterile, preservative‑free, clear to opalescent, colorless to slightly yellow, solution for intravenous use. SAPHNELO contains anifrolumab-fnia at a concentration of 150 mg/mL in a single-dose vial.
Each vial contains 300 mg (150 mg/mL) of anifrolumab-fnia, L-histidine (3 mg), L-histidine hydrochloride monohydrate (6 mg), L-lysine hydrochloride (18 mg), polysorbate 80 (1 mg), trehalose dihydrate (98 mg), and Water for Injection, USP. The pH is 5.9.
7CLINICAL STUDIES
The safety and efficacy of SAPHNELO were evaluated in three 52-week treatment period, multicenter, randomized, double-blind, placebo-controlled studies (Trial 1 [NCT01438489], Trial 2 [NCT02446912] and Trial 3 [NCT02446899]). Patients were diagnosed with SLE according to the American College of Rheumatology (1982 revised) classification criteria. All patients were ≥18 years of age and had moderate to severe disease, with a SLE Disease Activity Index 2000 (SLEDAI-2K) score ≥6 points, organ level involvement based on BILAG assessment, and a Physician’s Global Assessment [PGA] score ≥1, despite receiving standard SLE therapy consisting of either one or any combination of oral corticosteroids (OCS), antimalarials and/or immunosuppressants at baseline. Patients continued to receive their existing SLE therapy at stable doses during the clinical trials, with the exception of OCS (prednisone or equivalent) where tapering was a component of the protocol. Patients who had severe active lupus nephritis and patients who had severe active central nervous system lupus were excluded. The use of other biologic agents and cyclophosphamide were not permitted during the trials; patients receiving other biologic therapies were required to complete a wash-out period of at least 5 half‑lives prior to enrollment. All three studies were conducted in North America, Europe, South America and Asia. Patients received anifrolumab-fnia or placebo, administered by intravenous infusion, every 4 weeks.
Efficacy of SAPHNELO was established based on assessment of clinical response using the composite endpoints, the British Isles Lupus Assessment Group based Composite Lupus Assessment (BICLA) and the SLE Responder Index (SRI‑4).
BICLA response at Week 52, was defined as improvement in all organ domains with moderate or severe activity at baseline:
- Reduction of all baseline BILAG A to B/C/D and baseline BILAG B to C/D, and no BILAG worsening in other organ systems, as defined by ≥1 new BILAG A or ≥2 new BILAG B;
- No worsening from baseline in SLEDAI-2K, where worsening is defined as an increase from baseline of >0 points in SLEDAI-2K;
- No worsening from baseline in patients’ lupus disease activity, where worsening is defined by an increase ≥0.30 points on a 3-point PGA VAS;
- No discontinuation of treatment;
- No use of restricted medication beyond the protocol-allowed threshold.
SRI‑4 response, was defined as meeting each of the following criteria at Week 52 compared with baseline:
- Reduction from baseline of ≥4 points in the SLEDAI-2K;
- No new organ system affected as defined by 1 or more BILAG A or 2 or more BILAG B items compared to baseline;
- No worsening from baseline in the patients’ lupus disease activity defined by an increase ≥0.30 points on a 3‑point PGA visual analogue scale (VAS);
- No discontinuation of treatment;
- No use of restricted medication beyond the protocol-allowed threshold.
Trial 1 randomized 305 patients (1:1:1) who received anifrolumab-fnia, 300 mg or 1000 mg, or placebo for up to 52 weeks. The primary endpoint was a combined assessment of the SRI-4 and the sustained reduction in OCS (<10 mg/day and ≤OCS dose at week 1, sustained for 12 weeks) measured at Week 24.
Trial 2 and 3 were similar in design. Trial 2 randomized 457 patients who received anifrolumab-fnia 150 mg, 300 mg or placebo (1:2:2). Trial 3 randomized 362 patients (1:1) who received anifrolumab-fnia 300 mg or placebo. The primary endpoints were improvement in disease activity evaluated at 52 weeks, measured by SRI‑4 in Trial 2 and BICLA in Trial 3 (defined above). The common secondary efficacy endpoints included in both studies were the maintenance of OCS reduction, improvement in cutaneous SLE activity, and flare rate. During Weeks 8-40, patients with a baseline OCS ≥10 mg/day were required to taper their OCS dose to ≤7.5 mg/day, unless there was worsening of disease activity. Both studies evaluated the efficacy of anifrolumab-fnia 300 mg versus placebo; a dose of 150 mg was also evaluated for dose-response in Trial 2.
Patient demographics and disease characteristics were generally similar and balanced across treatment arms (Table 2).
Randomization was stratified by disease severity (SLEDAI-2K score at baseline, <10 vs ≥10 points), OCS dose on Day 1 (<10 mg/day vs ≥10 mg/day prednisone or equivalent) and interferon gene signature test results (high vs low).
The reduction in disease activity seen in the BICLA and SRI-4 was related primarily to improvement in the mucocutaneous and musculoskeletal organ systems. Flare rate was reduced in patients receiving SAPHNELO compared to patients who received placebo although the difference was not statistically significant.
BICLAresponder analysis: BICLA was the primary endpoint in Trial 3; anifrolumab-fnia 300 mg demonstrated statistically significant and clinically meaningful efficacy in overall disease activity compared with placebo, with greater improvements in all components of the composite endpoint. In Trial 1 and 2 BICLA was a pre-specified analysis. The BICLA results are presented in Table 3.
In Trial 3, examination of subgroups by age, race, gender, ethnicity, disease severity [SLEDAI-2K at baseline], and baseline OCS use did not identify differences in response to anifrolumab-fnia.
Figure 1 shows the proportion of BICLA responders through the 52-week treatment period in Trial 3.
Figure 1 Trial 3: Proportion (%) of BICLA Responders by Visit
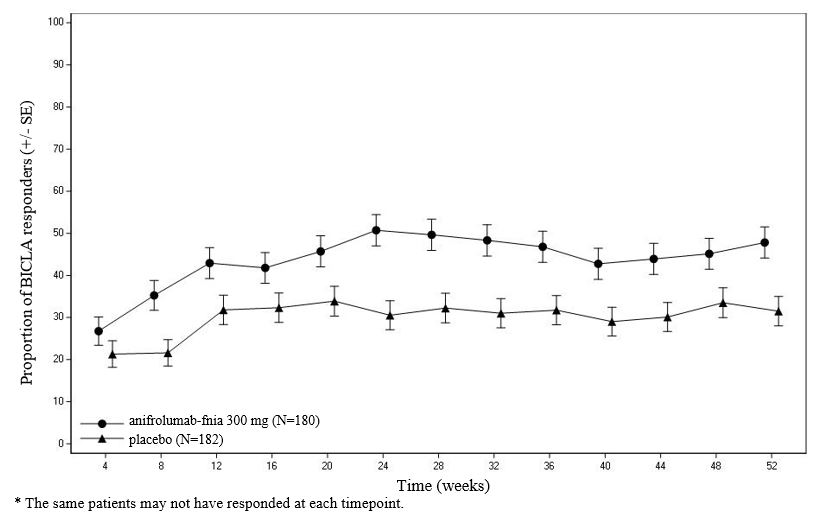
SRI-4 responder analysis: SRI-4 was the primary endpoint in Trial 2; treatment with anifrolumab-fnia did not result in statistically significant improvements over placebo. In Trials 1 and 3, SRI-4 was a pre-specified analysis. The SRI-4 results are presented in Table 4.
Effect on Concomitant Steroid Treatment: In Trial 3, among the 47% of patients with a baseline OCS use ≥10 mg/day, anifrolumab-fnia demonstrated a statistically significant difference in the proportion of patients able to reduce OCS use by at least 25% to ≤7.5 mg/day at Week 40 and maintain the reduction through Week 52 (p-value = 0.004); 52% (45/87) of patients in the anifrolumab-fnia group versus 30% (25/83) in the placebo achieved this level of steroid reduction (difference 21% [95% CI 6.8, 35.7]). Consistent trends in favor of anifrolumab-fnia compared to placebo, on effect of reduction of OCS use, were observed in Trial 1 and 2, but the difference was not statistically significant.
8HOW SUPPLIED/STORAGE AND HANDLING
SAPHNELO (anifrolumab-fnia) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow solution for intravenous infusion. It is packaged in a 2 mL clear glass vial containing 300 mg/2 mL (150 mg/mL) of anifrolumab-fnia.
SAPHNELO is available in a carton containing one single-dose vial (NDC-0310-3040-00).
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
Do not freeze. Do not shake.
9PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Serious Infections
Inform patients that SAPHNELO may decrease their ability to fight infections, and that serious infections, including fatal ones, occurred in patients receiving SAPHNELO in clinical trials. Also inform patients that they are at increased risk of respiratory infections and herpes zoster during treatment with SAPHNELO
Hypersensitivity Reactions/Anaphylaxis
Inform patients that serious hypersensitivity reactions, including anaphylaxis, have been reported in patients who received SAPHNELO. Instruct patients to immediately tell their healthcare provider or go to the emergency department of their nearest hospital, if they experience symptoms of an allergic reaction (e.g., anaphylaxis) during or after the administration of SAPHNELO
Immunizations
Inform patients that they should not receive live or live-attenuated vaccines while receiving SAPHNELO. Advise patients to discuss with their healthcare provider before seeking immunizations on their own
Pregnancy
Advise female patients to inform their healthcare provider if they intend to become pregnant during therapy, suspect they are pregnant or become pregnant while receiving SAPHNELO
Inform women that they can find information about a pregnancy exposure registry which monitors pregnancy outcomes in women exposed to SAPHNELO by calling AstraZeneca at 1-877-693-9268.
Manufactured by: AstraZeneca AB Södertälje, Sweden SE-15185
US License No. 2059
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2024
10PACKAGE/LABEL PRINCIPAL DISPLAY PANEL – 300mg/2mL (150 mg/mL)
NDC 0310-3040-00
SAPHNELO
(anifrolumab-fnia)
Injection
300 mg/2 mL
(150 mg/mL)
For Intravenous Infusion After Dilution
Must dilute before use. See prescribing information.
Single-dose vial. Discard unused portion.
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Keep vial in original carton to protect from light.
Do not shake or freeze.
1 single-dose vial
AstraZeneca
